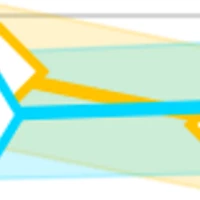
Bayesian active learning for optimization and uncertainty quantification in protein docking
Motivation: Ab initio protein docking represents a major challenge for optimizing a noisy and costly "black box"-like function in a high-dimensional space. Despite progress in this field, there is no docking method available for rigorous uncertainty quantification (UQ) of its solution quality (e.g. interface RMSD or iRMSD). Results: We introduce a novel algorithm, Bayesian Active Learning (BAL), for optimization and UQ of such black-box functions and flexible protein docking. BAL directly models the posterior distribution of the global optimum (or native structures for protein docking) with active sampling and posterior estimation iteratively feeding each other. Furthermore, we use complex normal modes to represent a homogeneous Euclidean conformation space suitable for high-dimension optimization and construct funnel-like energy models for encounter complexes. Over a protein docking benchmark set and a CAPRI set including homology docking, we establish that BAL significantly improve against both starting points by rigid docking and refinements by particle swarm optimization, providing for one third targets a top-3 near-native prediction. BAL also generates tight confidence intervals with half range around 25% of iRMSD and confidence level at 85%. Its estimated probability of a prediction being native or not achieves binary classification AUROC at 0.93 and AUPRC over 0.60 (compared to 0.14 by chance); and also found to help ranking predictions. To the best of our knowledge, this study represents the first uncertainty quantification solution for protein docking, with theoretical rigor and comprehensive assessment. Source codes are available at https://github.com/Shen-Lab/BAL.